 CALL US NOW:
CALL US NOW: 












 March 21, 2022
March 21, 2022  John Mwesigwa
John Mwesigwa Carnitine Palmitoyltransferase II Deficiency – Lipid Metabolism Disorders
Metabolism is the process your body uses to make energy from the food you eat. Food is made up of proteins, carbohydrates, and fats. Chemicals in your digestive system (enzymes) break the food parts down into sugars and acids, your body’s fuel. Your body can use this fuel right away, or it can store the energy in your body tissues. If you have a metabolic disorder, something goes wrong with this process.
Lipid metabolism disorders, such as Gaucher disease and Tay-Sachs disease, involve lipids. Lipids are fats or fat-like substances. They include oils, fatty acids, waxes, and cholesterol. If you have one of these disorders, you may not have enough enzymes to break down lipids. Or the enzymes may not work properly and your body can’t convert the fats into energy. They cause a harmful amount of lipids to build up in your body. Over time, that can damage your cells and tissues, especially in the brain, peripheral nervous system, liver, spleen, and bone marrow. Many of these disorders can be very serious, or sometimes even fatal.
These disorders are inherited. Newborn babies get screened for some of them, using blood tests. If there is a family history of one of these disorders, parents can get genetic testing to see whether they carry the gene. Other genetic tests can tell whether the fetus has the disorder or carries the gene for the disorder.
Enzyme replacement therapies can help with a few of these disorders. For others, there is no treatment. Medicines, blood transfusions, and other procedures may help with complications.
Carnitine palmitoyltransferase II (CPT II) deficiency is a condition that prevents the body from using certain fats for energy, particularly during periods without food (fasting). There are three main types of CPT II deficiency: a lethal neonatal form, a severe infantile hepatocardiomuscular form, and a myopathic form.The lethal neonatal form of CPT II deficiency becomes apparent soon after birth. Infants with this form of the disorder develop respiratory failure, seizures, liver failure, a weakened heart muscle (cardiomyopathy), and an irregular heart beat (arrhythmia). Affected individuals also have low blood sugar (hypoglycemia) and a low level of ketones, which are produced during the breakdown of fats and used for energy. Together these signs are called hypoketotic hypoglycemia. In many cases, the brain and kidneys are also structurally abnormal. Infants with the lethal neonatal form of CPT II deficiency usually live for a few days to a few months.The severe infantile hepatocardiomuscular form of CPT II deficiency affects the liver, heart, and muscles. Signs and symptoms usually appear within the first year of life. This form involves recurring episodes of hypoketotic hypoglycemia, seizures, an enlarged liver (hepatomegaly), cardiomyopathy, and arrhythmia. Problems related to this form of CPT II deficiency can be triggered by periods of fasting or by illnesses such as viral infections. Individuals with the severe infantile hepatocardiomuscular form of CPT II deficiency are at risk for liver failure, nervous system damage, coma, and sudden death.The myopathic form is the least severe type of CPT II deficiency. This form is characterized by recurrent episodes of muscle pain (myalgia) and weakness and is associated with the breakdown of muscle tissue (rhabdomyolysis). The destruction of muscle tissue releases a protein called myoglobin, which is processed by the kidneys and released in the urine (myoglobinuria). Myoglobin causes the urine to be red or brown. This protein can also damage the kidneys, in some cases leading to life-threatening kidney failure. Episodes of myalgia and rhabdomyolysis may be triggered by exercise, stress, exposure to extreme temperatures, infections, or fasting. The first episode usually occurs during childhood or adolescence. Most people with the myopathic form of CPT II deficiency have no signs or symptoms of the disorder between episodes.
-
 A1CNow Self Check
$80.00
A1CNow Self Check
$80.00
-
 G.LAB Digital Automatic Blood Pressure Monitor, 2.3 Count
$23.99 — available on subscription
G.LAB Digital Automatic Blood Pressure Monitor, 2.3 Count
$23.99 — available on subscription
-
 D-Mannose Vitamins
$29.97 — available on subscription
D-Mannose Vitamins
$29.97 — available on subscription
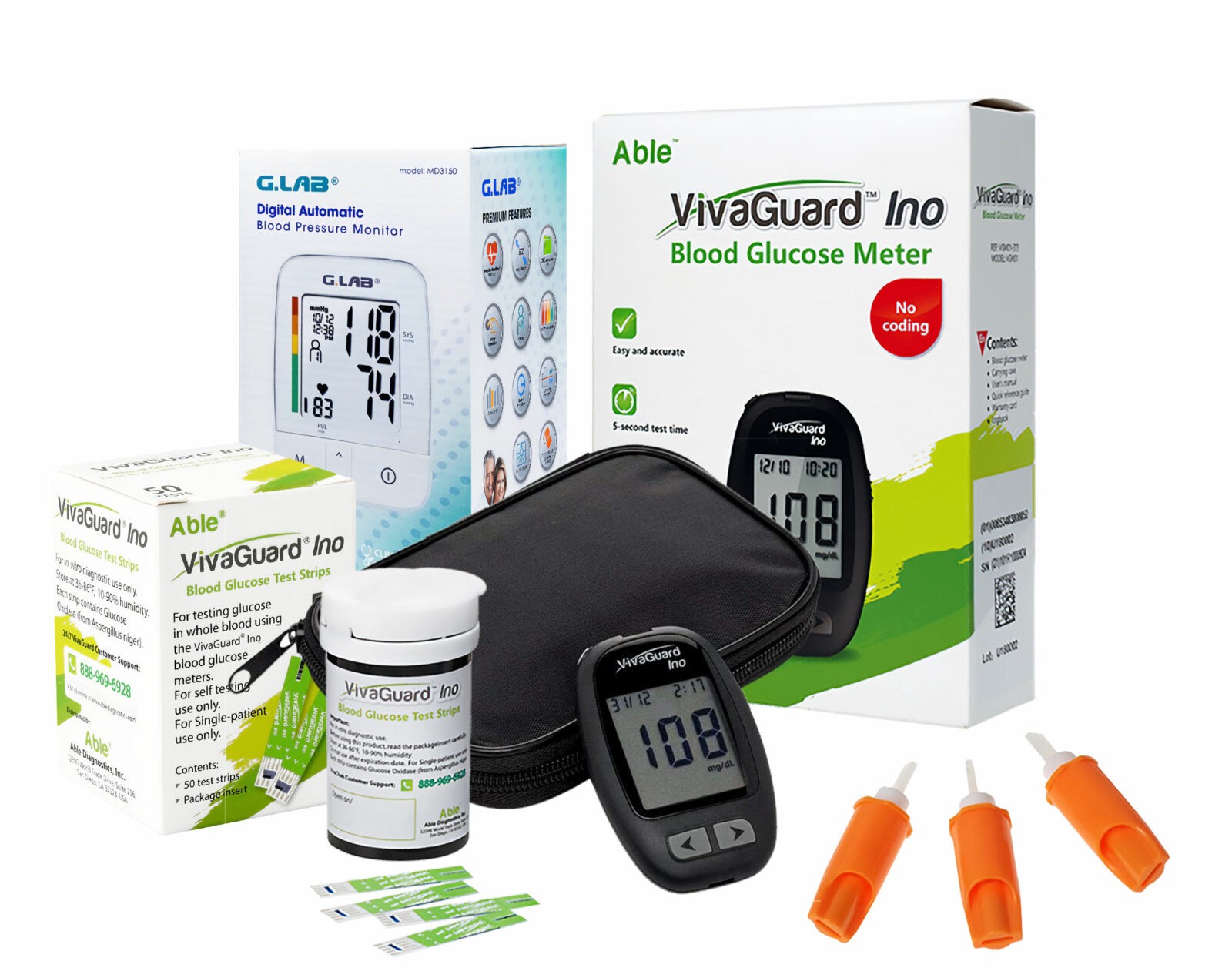
Get Testing Supplies from $8/Month
Glucose Meter
FDA-approved. Results in 4 seconds. Requires tiny
0.4
µL
sample
size.
Control Solution
Perfect tool to ensure test strips are providing
accurate
results.
Test Strips
Accurate test strips that support re-dosing.
Lancing Device
Five depth settings for maximum comfort
Lancets
Universal design that is compatible with most
lancing
devices.
Carrying Case
High-quality materials to store and protect
testing
supplies.